
데이터 분석에서 많은 양의 데이터를 처리할 때, 데이터의 차원을 축소하고 중요한 특성을 선택하는 것은 매우 중요한 작업이다. 이를 통해 모델의 복잡성을 줄이고 계산 효율성을 향상시키며, 더 나은 모델 성능을 달성할 수 있기 때문이다. 다루어야 할 내용이 적지 않기 때문에, 본 글에서는 '데이터 차원 축소'에 대해서 우선 다루겠다. 이후의 글에서는 데이터 정리의 다른 방법인 '특성 선택'에 대해서 적어보겠다.
데이터 차원 축소 기술 (Dimensionality Reduction Techniques)
아래 표는 대표적인 5가지 데이터 차원 축소 방법에 대한 요약이다.
| 기술 | 설명 | 필요성 | 예시 |
| PCA (주성분 분석) |
고차원 데이터의 주요 특성을 추출하여 데이터를 낮은 차원으로 투영하는 기술 | 데이터의 차원을 줄여 계산 비용을 절감하고 데이터 시각화와 분석을 용이하게 함 | 이미지 압축, 얼굴 인식, 유전자 발현 데이터 분석 |
| SVD (특이값 분해) |
특이값 분해를 통해 행렬을 세 개의 행렬로 분해하는 기술 | 대규모 데이터의 차원을 줄이고 행렬의 특성을 추출함 | 자연어 처리, 이미지 압축, 추천 시스템 |
| t-SNE (t-분포 확률적 임베딩) |
고차원 데이터를 저차원 공간에 매핑하여 데이터 간의 유사성을 보존하는 기술 | 데이터의 시각화와 군집화에 활용되며, 비선형 관계를 파악하는 데 유용함 | 고차원 데이터의 시각화, 클러스터링, 특징 추출 |
| LDA (잠재 디리클레 할당) |
문서 집합 내의 숨겨진 토픽을 추론하고 각 문서의 토픽 분포를 파악하는 확률적 생성 모델 | 문서 간의 유사성을 파악하고, 토픽을 이해하여 문서의 구조를 분석함 | 텍스트 마이닝, 문서 분류, 토픽 모델링 |
| NMF (비음수 행렬 분해) |
행렬을 두 개 이상의 비음수 행렬로 분해하여 데이터의 특성을 추출하는 기술 | 데이터의 차원을 줄이고, 중요한 특성을 추출하여 분석함 | 이미지 처리, 음성 처리, 텍스트 마이닝 |
각각에 대해서 하나씩 설명하면 아래와 같다.
주성분 분석 (PCA, Principle Component Analysis)
주성분 분석(Principal Component Analysis, PCA)은 데이터 분석에서 자주 등장하는 개념이다. 혹시 영어가 많이 부담이 되지 않는다면, Josh Stammer의 StatQuest 채널에 있는 PCA에 대한 설명을 참조하기 바란다. StatQuest: PCA Step-by-Step
주성분 분석이 필요한 이유는 데이터의 차원을 줄이고 중요한 정보를 더 효율적으로 추출하기 위해서이다. 우리가 다루는 데이터는 종종 매우 복잡하고 다차원일 수 있다. 이러한 다차원 데이터를 분석하려면 데이터를 이해하기 쉬운 형태로 변환하는 것이 중요하며, 그 중 가장 대표적인 기법이 주성분 분석이다.
주성분 분석은 데이터의 주요 특성을 추출하여 데이터를 저차원으로 투영한다. 여기서 추출되는 주요 특성을 '주성분(Principle Component)'이라고 한다. 주성분 분석을 통해 우리는 데이터의 분산을 최대한 보존하는 새로운 축을 찾을 수 있고, 이러한 새로운 축은 데이터를 더 쉽게 이해할 수 있도록 도와준다.
주성분 분석을 수행한 후에는 회귀분석이나 클러스터링 등의 머신러닝 기법들을 사용해서 더 심화된 데이터 분석을 할 수 있게 해 준다.
아래는 주성분 분석을 하는 간단한 예제이다. 'prcomp()'함수를 사용하여 주요 성분을 찾아내고, 주요 성분은 'pc1', 'pc2' 등에 저장해 준다.
# Load the iris dataset
data(iris)
# Subset the dataset to include only numeric columns
iris_numeric <- iris[, sapply(iris, is.numeric)]
# Scale the numeric features
scaled_iris <- scale(iris_numeric)
# Perform PCA
pca_result <- prcomp(scaled_iris)
# Extract the principal components
pc1 <- pca_result$x[, 1]
pc2 <- pca_result$x[, 2]
# Plot the data points in the new feature space
plot(pc1, pc2, col = iris$Species, pch = 16, main = "PCA of Iris Dataset")
legend("topright", legend = levels(iris$Species), col = 1:3, pch = 16)
특이값 분해 (SVD, Singular Value Decomposition)
SVD는 PCA와 밀접한 관련이 있는 기술로, 행렬을 세 부분으로 분해하여 데이터를 축소합니다.
특이값 분해(Singular Value Decomposition, SVD)는 데이터 분석과 차원 축소에 널리 사용되는 중요한 도구이다. 특이값 분해는 데이터를 세 개의 행렬의 곱으로 분해함으로써 데이터를 축소한다. 이러한 세 개의 행렬은 각각 데이터의 특이값, 왼쪽 특이 벡터, 오른쪽 특이 벡터라고 불리며, 각 행렬에는 주요 특성이 담겨 있기 때문에 개념상으로 주성분 분석(PCA)과도 밀접한 관련이 있다고 할 수 있다.
주성분 분석에서와 마찬가지로 매우 대략적인 사용 예시는 아래와 같을 수 있다. iris 데이터 세트에서 추출한 'iris_numeric' (위 '주성분 분석' 예시 R 코딩 참조)에 'svd()'함수를 사용하면 특이값이 r, u, v로 분해되어 저장된다.
# Perform Singular Value Decomposition
svd_result <- svd(iris_numeric)
# Extract the singular values
singular_values <- svd_result$d
# Print the singular values
print(singular_values)
# Compute the rank of the matrix
rank_matrix <- sum(singular_values > 0)
# Print the rank of the matrix
print(rank_matrix)
# Compute the left singular vectors (U)
left_singular_vectors <- svd_result$u
# Compute the right singular vectors (V)
right_singular_vectors <- svd_result$v
# Compute the matrix approximation using the reduced rank
reconstructed_matrix <- left_singular_vectors[, 1:rank_matrix]
%*% diag(singular_values[1:rank_matrix])
%*% t(right_singular_vectors[, 1:rank_matrix])
t-분산 확률적 이웃 임베딩 (t-SNE, t-distributed stochastic neighbor embedding)
t-분산 확률 인접성 임베딩 (또는 t-분포 확률적 임베딩) (t-distributed Stochastic Neighbor Embedding, t-SNE)는 고차원 데이터를 시각화하기 방법으로 사용된다. 이 기술은 이해하기 힘든 데이터를 저차원 공간으로 변환하여, 그 데이터의 복잡한 구조를 시각적으로 이해할 수 있도록 도와준다.
t-SNE는 주어진 데이터의 각 점 사이의 거리를 확률 분포로 변환한 후, 저차원 공간에서도 가능한 유사한 확률 분포를 유지해 준다. 이러한 방법을 통해서 저차원에서도 고차원 데이터의 구조를 유지할 수 있다. 실제로, t-SNE는 다양한 분야에서 널리 사용되고 있다. 예를 들어, 이미지 인식, 자연어 처리, 유전자 데이터 분석 등에서 t-SNE를 사용하여 데이터를 시각화하고 분석하는 경우가 많다.
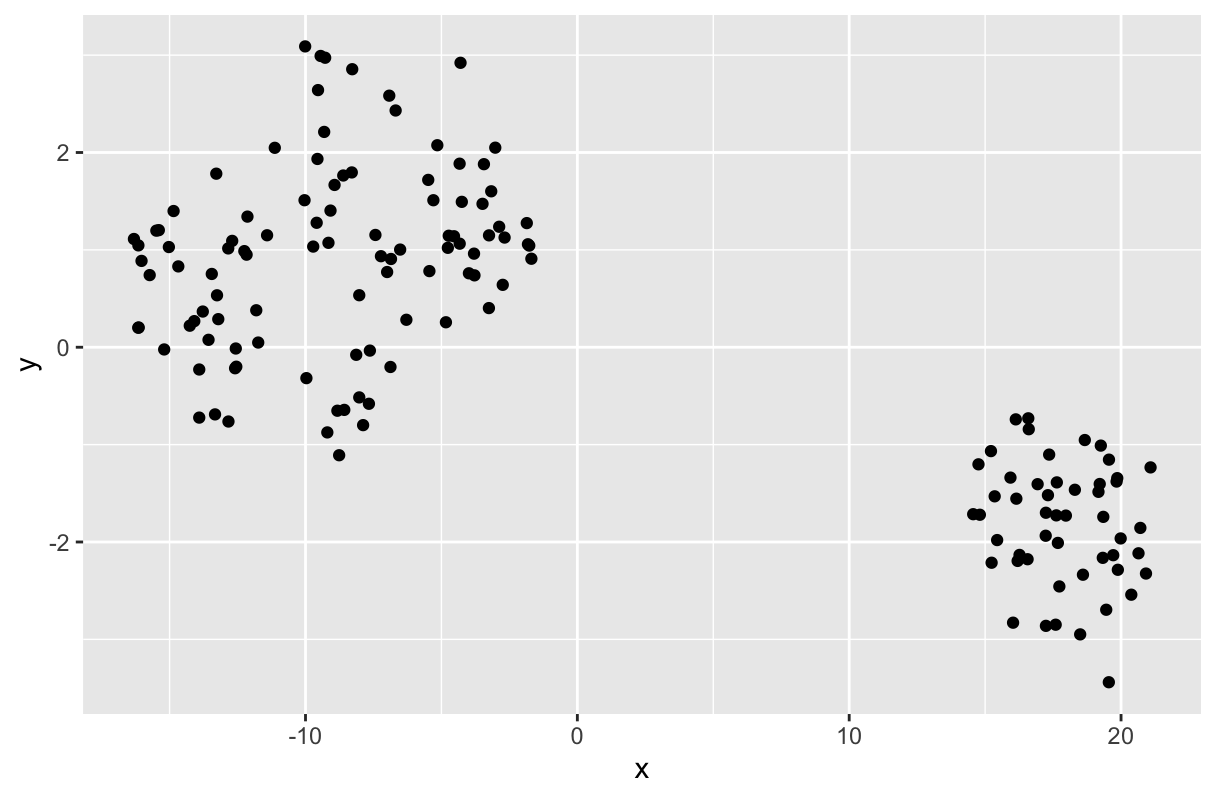
t-SNE를 사용하기 위해서는 'Rtsne' 패키지가 설치해야 한다. 아래는 예도 마찬가지로 iris 데이터 세트를 사용하였다. 해당 데이터 세트의 첫 4개 항목은 'Sepal.Length', 'Sepal.Width', 'Petal.Length', 'Petal.Width'로 5번째 항목인 iris 꽃의 종에 따라 수집된 꽃잎의 길이와 너비값들이다. 원래 데이터인 iris를 보면 알겠지만, 패턴이 전혀 눈에 들어오지 않는다. 하지만 t-SNE를 수행한 후 그래프를 그리면, 각 집단의 특성에 따라 구분이 된 채로 그려진다.
# Install Rtsne Package and load the library
install.packages("Rtsne")
library(Rtsne)
library(tidyverse)
# Load the iris dataset
data(iris)
# Subset the dataset to include only numeric columns
iris_numeric <- iris[, sapply(iris, is.numeric)]
# Forming the Matrix using unique values in iris for the first 4 columns only
iris_matrix <- as.matrix(iris_numeric[,1:4])
# Perform t-SNE
tsne_result <- tsne(iris_numeric)
# Make matrix to Dataframe
tsne_df <- data.frame(x= tsne_result$Y[,1],
y= tsne_result$Y[,2])
# Plot the t-SNE data frame
ggplot(tsne_df, label = Species) +
geom_point(aes(x=x, y=y))
잠재 디리클레 할당 (LDA, Latent Dirichlet Allocation)
잠재 디리클레 할당(Latent Dirichlet Allocation, LDA)은 토픽 모델링을 위한 확률적 생성 모델이다. 이 기술은 문서 집합 내에서 토픽을 추론하고 각 문서의 토픽 분포를 파악하는 데 사용된다. 이런 정의만으로는 감이 쉽게 오지 않을 것이다. 토픽은 무엇이고, 왜 토픽을 추론해 내야 하는가.
이를 위해서 우선, LDA가 텍스트마이닝에서 주로 사용되는 기법이라는 점을 먼저 알아야 한다. 토픽은 추출해 낸 텍스트의 공통된 주제이다. 즉, 여러 단어들을 뽑아낸 후에, 토픽(주제)에 맞게 카테고리화된다. 텍스트마이닝된 빅데이터를 효과적으로 분석하려면 토픽과 문서 간의 관계를 이해할 수 있어야 한다. LDA는 이러한 목적을 달성하기 위해 개발된 기술인 것이다.
LDA는 주어진 문서 집합을 설명할 수 있는 토픽들의 확률 분포를 추정한다. 이를 통해 각 문서의 토픽 분포를 파악할 수 있으며, 이를 기반으로 문서 간의 유사성을 계산해 낸다. 실제로, LDA는 다양한 분야에서 널리 사용되고 있다. 예를 들어, 웹 문서의 토픽 모델링, 고객 리뷰의 감성 분석, 뉴스 기사의 주제 분류 등에 활용될 수 있다.
아래는 R에서 제공하는 토픽모델 패키지에 있는 'AssociatedPress'라는 텍스트 데이터를 사용한 LDA예제이다. 'corpus'는 텍스트마이닝된 단어들의 '말뭉치'이다.
# Load the required libraries
#install.packages("tm")
#install.packages("topicmodels")
library(tm)
library(topicmodels)
# Load the AssociatedPress corpus
data("AssociatedPress")
# Preprocess the corpus
corpus <- tm_map(AssociatedPress, content_transformer(tolower))
corpus <- tm_map(corpus, removePunctuation)
corpus <- tm_map(corpus, removeNumbers)
corpus <- tm_map(corpus, removeWords, stopwords("en"))
corpus <- tm_map(corpus, stripWhitespace)
# Create a document-term matrix
dtm <- DocumentTermMatrix(corpus)
# Perform LDA
lda_model <- LDA(dtm, k = 3)
# Print the topics
topics(lda_model)
비음수 행렬 분해 (NMF, Non-negative MAtrix Factorization)
비음수 행렬 인수분해(Non-negative Matrix Factorization, NMF)는 행렬을 두 개 이상의 비음수 행렬로 분해하는 기술이다. 이 기술은 이미지 처리에서 이미지의 특성을 추출하거나, 음성 처리에서 음성 신호의 특성을 분석하는 등 다양한 응용 분야에서 사용될 수 있다.
NMF는 주어진 행렬을 두 개 이상의 비음수 행렬로 분해하여 원본 데이터의 특성을 잘 나타내는 요인들을 추출한다. 이를 통해 데이터의 차원을 줄이고, 중요한 패턴이나 구조를 포착할 수 있다. 또한, NMF는 비음수 제약을 사용하여 더 의미 있는 요인을 추출할 수 있다.
아래는 'NMF'패키지를 사용한 예제이다.
# Load the required library
#install.packages("jpeg")
#install.packages("NMF")
library(jpeg)
library(NMF)
# Load and display the image
img <- readJPEG("your_image.jpg")
plot(as.raster(img))
# Convert the image to grayscale
gray_img <- rgb2gray(img)
# Reshape the image matrix
img_matrix <- matrix(gray_img, ncol = ncol(gray_img))
# Perform NMF
nmf_result <- nmf(img_matrix, 3)
# Reconstruct the image using the basis matrix and coefficients
reconstructed_img <- nmf_result$W %*% nmf_result$H
# Plot the reconstructed image
plot(as.raster(reconstructed_img))참고: 2024년 4월 현재, 'NMF' 패키지 설치를 위한 'Biobase' 패키지가 존재하지 않는 것으로 나온다. 추후에 원인을 파악해서 글을 수정하도록 하겠다.
'데이터 기본' 카테고리의 다른 글
| 데이터 변환: 정규화, 표준화 및 집계 기술 (0) | 2024.04.09 |
|---|---|
| 데이터 정제: 중복 제거, 누락 값 처리, 이상 값 처리 기술 (0) | 2024.04.06 |
| K-폴드 교차검증법: 움파룸파의 "A thousand fold!!" (0) | 2024.03.11 |
| 데이터 샘플링: 왜, 언제 해야 하는가 (0) | 2023.02.27 |
| 데이터 샘플링: 통계적 및 비통계적 샘플링 방법 (0) | 2023.02.20 |



